Introduction
In this section we will cover:
- What is long range sequencing?
- SMRT sequencing
- Nanopore sequencing
- Different ONT sequencers
- Advantages of long-read sequencing
- Disadvantages of long-read sequencing
- A standard workflow
1. What is long read sequencing?
The genomes of many organisms are far too big to be sequenced in one long continuous string.

In order to sequence these massive genomes, short-read sequencing was developed. Here DNA is broken into millions of tiny fragments that are amplified (copied) and then sequenced.
This process produces millions of short sequences or “reads” which are typically 75-300bp in length which are then pieced back together computationally.
Short-read workflow

In contrast, long-read sequencing allows us to retrieve much longer sequence reads that are around >6,000bp in length. Some long read sequencing systems have even produced reads that are over 2,000,000bp (2 MB) in length.
Additionally long-read methodologies typically sequence single molecules of DNA directly and in real-time, foregoing the need for amplification.
Long-read workflow

The two best known producers of ‘true’ long-read sequencing technologies are Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). Bother offer platforms that directly sequence single molecules of DNA or RNA in ‘real-time’ and which can produce significantly longer reads than current short-read platforms.
2. SMRT sequencing
Single Molecule, Real Time (SMRT) sequencing is the name of PacBio’s methodology.
SMRT sequencing workflow:
- DNA is extracted from the sample using standard methods.
- DNA is then fragmented.
- Sequencing adapters are ligated to the fragments to create circular templates.

- Primer and polymerase are then added.
- The library is then loaded into a “SMRT cell” containing millions of ‘wells’ called Zero Mode Wave-guides (ZMW’s).
- A single molecule of DNA is immobilised inside each ZMW.
- The sequencing reaction now takes place. As the DNA polymerase incorporates nucleotides light is emitted and measured.


PacBio SMRT sequencing has two modes:
- Circular Consensus Sequencing (CCS): Sequencies a smaller circular template over and over to produce a high quality read.
- Continuous Long Read Sequencing (CLR): Produces the longest read by sequencing long templates.
Original Publication: Eid, J., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science, 323(5910), 133–138.
———– Disclaimer ———–
We have never actually used or worked with PacBio sequencers/data. From now on this course will focus on Nanopore sequencing.
If you would like more information about PacBio, I suggest you contact Prof. Steven Marsh or Dr. James Robinson who are based at the Anthony Nolan Research Institute in London.
They have been using PacBio sequencing for HLA typing over the past few years and have expert knowledge on this system.
3. Nanopore sequencing
Nanopore sequencing is the general name for ONT’s methodology.
Nanopore sequencing workflow:
- DNA is extracted from the sample using standard methods.
- You now choose which type of sequencing you would like to do, below are two examples:
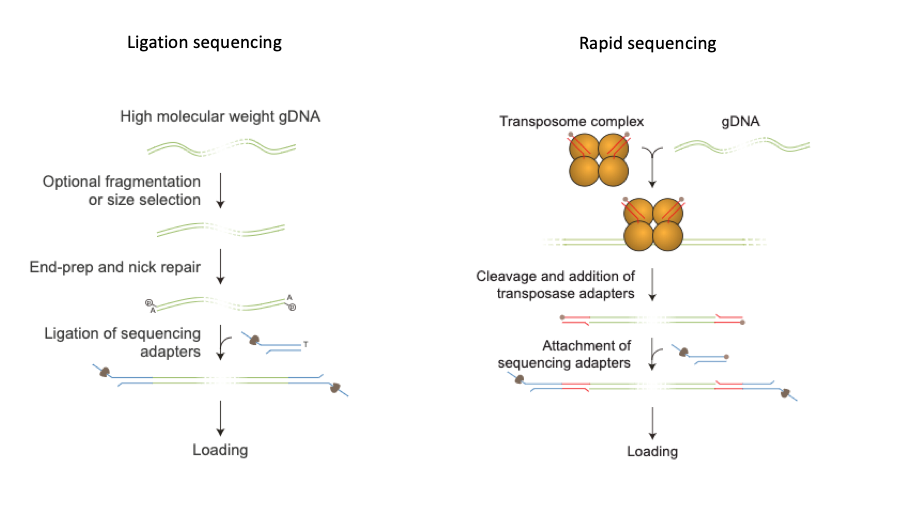
- Libraries are now loaded into flow cells for sequencing.
- Each flow cell contains an electrically-resistant membrane with hundreds of protein nanopores embedded within it.
- A voltage is applied across the membrane and an ionic current begins to flow through the nanopore (as the membrane is electrically resistant).
- As the DNA passes through the nanopore, the ionic current is disrupted and this event is recorded.
- The specific pattern of disruption is analysed to determine DNA sequence.


Note in the second image, that as DNA passes through the pore a motor protein unwinds it. This actually slows down the DNA passage enough to allow measurement of the ionic field disruption.
Further reading:
Branton, D. et al. (2008). The potential and challenges of nanopore sequencing. Nature Biotechnology, 26(10), 1146-1153.
Deamer, D., et al. (2016). Three decades of nanopore sequencing. Nature Biotechnology, 34(5), 518-24.
4. Different ONT sequencers
ONT offer various different sequencing machines - the biggest difference is sequencing throughput (how many bases you can sequence) and their size.
Below are examples of the most commonly used.

We have been lucky enough to use all three of these. The MinION and GridION are well suited to smaller genomes or targetted sequencing, the PromethION is better suited to WGS of large genomes such as human.
5. Advantages of long-read sequencing
Genome assembly: Reassembling large genomes from short reads can be challenging, as many fragments look highly similar without additional context. Long-read data can make this task simpler as the reads are more likely to look distinct, allowing them to be assembled together with less ambiguity and error.
Complex variant detection: Large and complex rearrangements, large insertions or deletions of DNA, repetitive regions, highly polymorphic regions, or regions with low DNA nucleotide diversity can be extremely hard to observe using short read sequencing. Long reads can span across larger parts of these regions, so are able to detect more of these variants, which may be clinically relevant.
Haplotype phasing: In clinical/medical genetics it is often critically important to determine the inheritance mode of multiple variants that may cause disease. Long reads are able to provide the long-range information for resolving haplotypes without additional statistical inference or parental sequencing.
Other applications: Long-read platforms can also directly sequence RNA with no requirement for amplification or reverse transcription allowing us to better characterise the transcriptome. We can also detect epigenetic modifications such as methylation status in the electrical disruption data, this can be done from a regular sequencing run!
6. Disadvantages of long-read sequencing
The biggest limitation for nanopore sequencing is that it has a much lower read accuracy than short read platforms. Nanopore sequencing is therefore sub-optimal for detections of single nucleotide variants (SNV’s) and small insertions and deletions (indels). High coverage of a given region is required to facilitate calling of these variants.
When homopolymer regions move through the pore with no change in current amplitude, the number of nucleotides associated with each detectable amplitude level must be inferred. This introduces random insertion or deletion errors.
However, the accuracy of nanopore sequencing is highly dependent on the specific protein nanopore used. The current R9.X pore can ‘read’ about 5 bases at a time, which is what makes homopolymer detection difficult. Recently, nanopore have released the R10.X pore which contains multiple readers, this means more base signal can be observed over a longer sequence distance.
This translates to an accuracy improvement from ~Q34 (error rate: 1 in 1,000) with R9.X pores to Q40 (error rate: 1 in 10,000) at 75x depth.

Karst, S., et al. (2020), Enabling high-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. bioRxiv.
Computational methods can also be used to improve overall consensus accuracy. These algorithms assess raw signal data to ‘polish’ a consensus sequence made using nanopore data. An example is Nanopolish.
7. A standard workflow
In this course we are going to take you through a standard Nanopore sequencing workflow. You can then modify this ‘vanilla’ pipeline to suit your own analysis.
